| Главная » Статьи » dermatoscop » Популярные статьи |
16.06.2014,
|
Томас Б. Фитцпатрик, Дэвид Б. Мошер (Thomas B. Fitzpatrick, David B. Mosher)
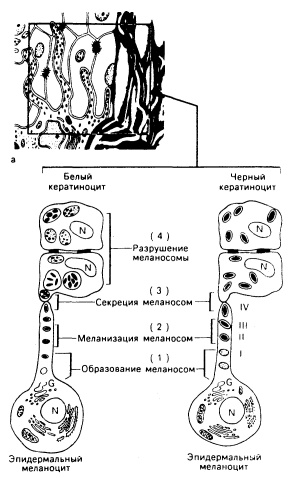
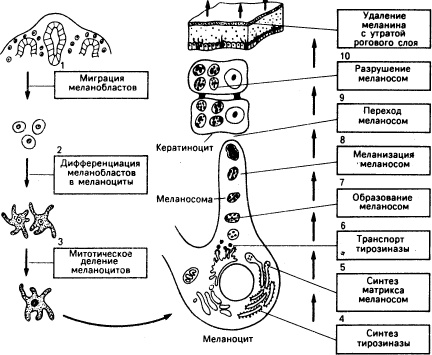
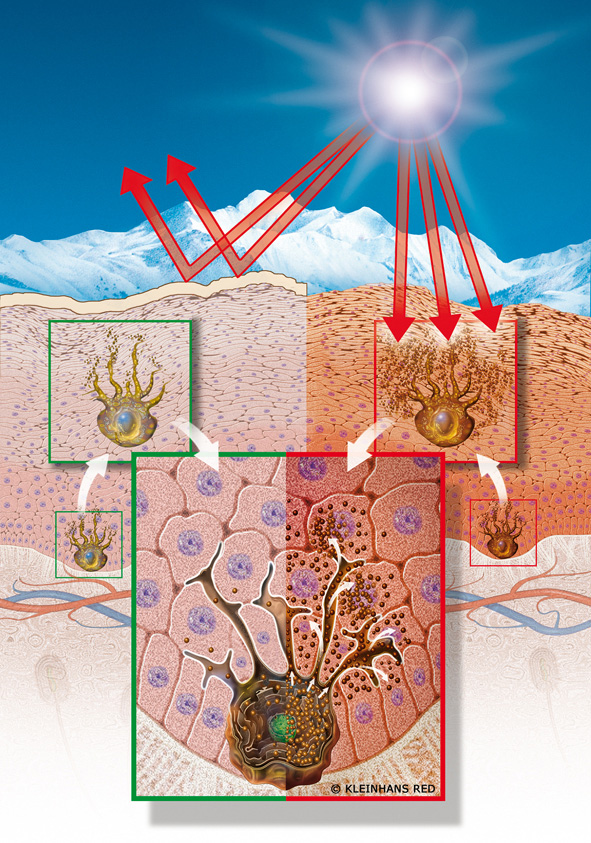
Рис. 51-2. Схематическое изображение эпидермального меланинового комплекса. а: Меланоцит снабжает меланосомами группу кератиноцитов. Представлены четыре биологических процесса в образовании и упаковке меланосом, лежащие в основе меланиновой пигментации у представителей негроидной и европеоидной популяций (б): 1) образование в меланоцитах меланосом; 2) меланизация меланосом в меланоцитах; 3) секреция кератиноцитами меланоцитов; 4) транспорт меланоцитов кератиноцитами с разрушением меланосом в лизосомоподобных органеллах (у представителей европеоидной популяции) или без заметного их разрушения (у представителей негроидной популяции). Обратите внимание на разные размеры меланосом в черных и белых эпидермальных кератиноцитах: в черных кератиноцитах они не образуют агрегатов, в белых — группы из нескольких меланосом образуют скопления в ограниченных мембраной лизосомоподобных органеллах: меланосомы часто фрагментированы (G — аппарат Гольджи. N — ядро, I-IV — четыре стадии развития меланосомы).
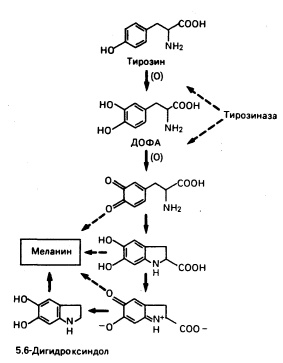
Рис. 51-3. Биосинтез меланина из тирозина.
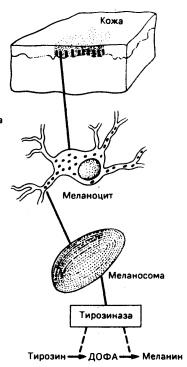
Рис. 51-4. Схематическое изображение меланогенеза в коже человека при световом и электронно-микроскопическом исследовании.
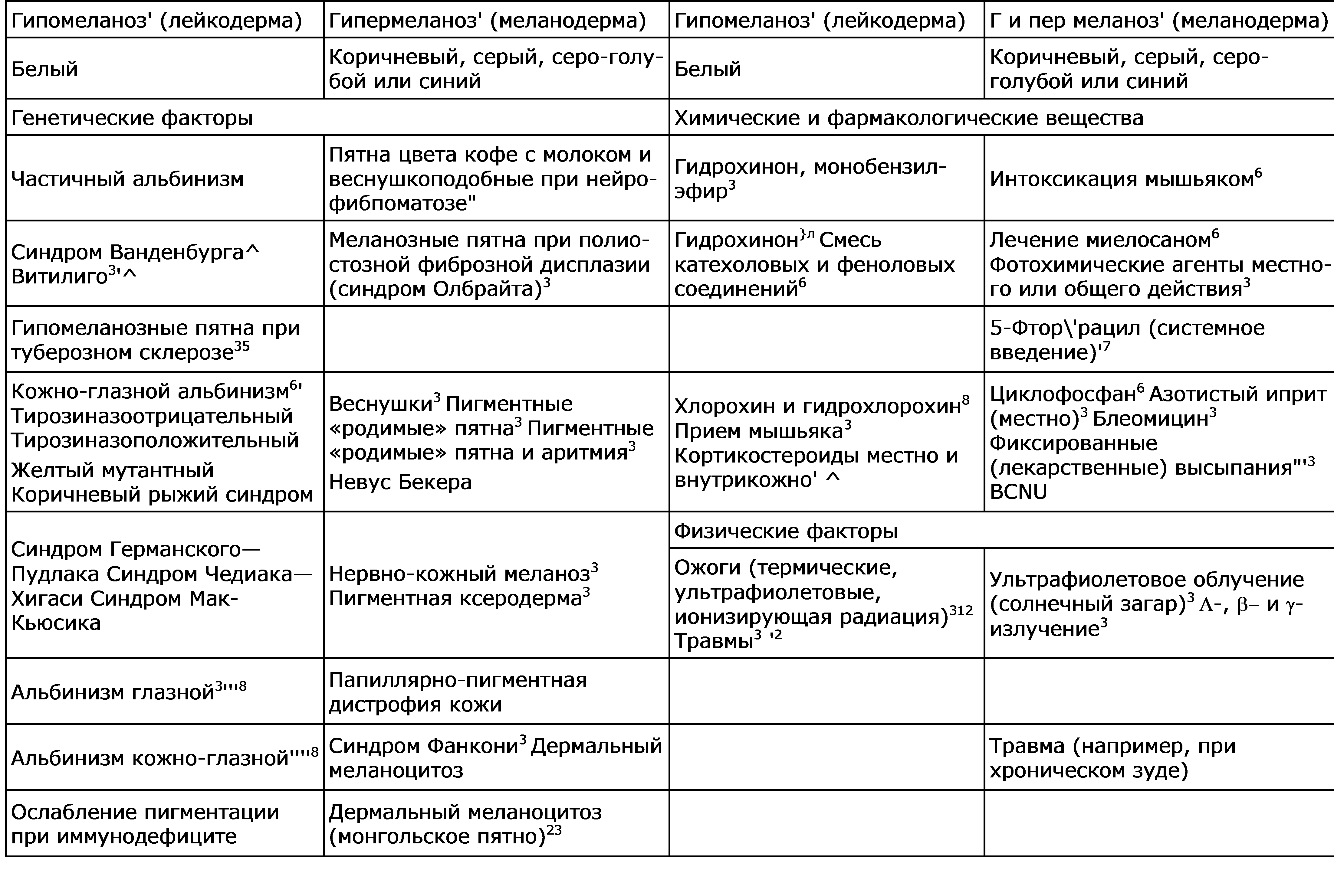
Рис. 51-5. Схематическое изображение морфологических и метаболических механизмов меланиновой пигментации эпидермиса. Нарушения системы меланоцитов: гипомеланозы и. гипермеланозы Таблица 51-1. Изменения пигментации как диагностические признаки в терапии
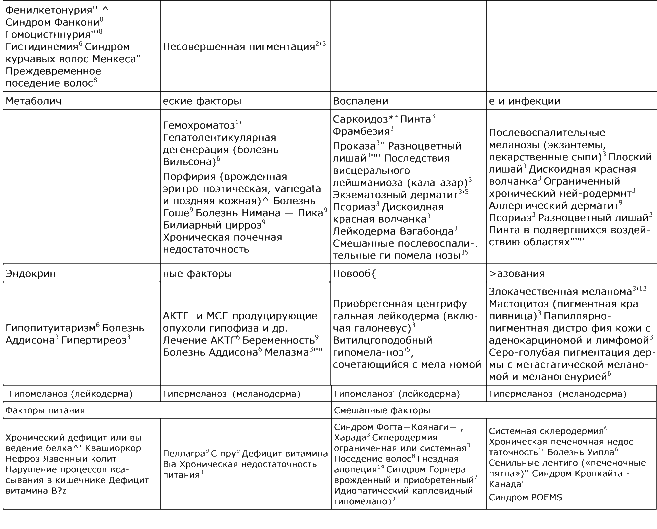
Таблица 51-2. Нарушение меланиновой пигментации
Распознавание отграниченного гипомеланоза (белые пятна), серого, серо-голубого или синего гипермеланоза обычно трудностей не вызывает. При слабо выраженном гипомеланозе у больного с очень светлой или незагорелой кожей ее изменения могут быть неотчетливыми, диагностике способствует исследование в темном свете (лампа Вуда), при котором усиливается контраст между участками кожи с измененной эпидермальной пигментацией и здоровой, но не усиливаются различия между здоровой кожей и зонами повышенной дермальной пигментации. Нередко бывает трудно дифференцировать диффузную коричневую гиперпигментацию (например, при болезни Аддисона) или диффузный гипомеланоз (при альбинизме) от нормальной пигментации из-за широкого спектра ее колебаний у здоровых. Диффузные изменения цвета кожи могут быть малозаметными, часто сам больной не подозревает о необычном, необъяснимом, постепенно прогрессирующем потемнении кожи типа сохраняющегося летнего загара. Степень гипермеланоза связана с исходным цветом кожи больного. При болезни Аддисона у выходца из Средиземноморья (например, жители Италии, Франции или Испании) кожа может оказаться интенсивно пигментированной, а у человека со светлой кожей может развиваться только минимальная степень гипермеланоза. Изменение пигментации в слизистых оболочках и некоторых зонах, например в подмышечных впадинах и на ладонных поверхностях, обычно определяется легче, чем генерализованная коричневая гиперпигментация. Генетические нарушения обмена меланина. Больные с нарушениями пигментации могут предъявить жалобы на потемнение кожи общее или пятнами, появление «белых» или «родимых пятен» (см. табл. 51-1), у других наступают глухота, ирит, появляются припадки, а изменения пигментации носят случайный характер. Приводимый далее анализ базируется вместе с тем скорее на этиологических факторах, нежели на симптоматологии. Кожно-глазной альбинизм относится к аутосомно-рецессивным признакам и характеризуется врожденным равномерным гипомеланозом кожи и волос. Случаи только кожного альбинизма не встречаются, но глазной альбинизм на фоне неизмененной или минимально измененной кожи был зарегистрирован. Классическими признаками при кожно-глазном альбинизме служат выраженный гипомеланоз или амеланоз кожи, белые или почти белые волосы, светобоязнь, нистагм, гипопигментированное глазное дно, просвечивающая радужка. Этот вид альбинизма может классифицироваться в зависимости от наличия или отсутствия тирозиназы в фолликулах вырванных волос волосистой части головы (реакция инкубации фолликула волоса). Волосяные луковицы волос у здорового человека темнеют при инкубации с тирозином. При кожно-глазном альбинизме они также могут иногда темнеть в этих условиях (тирозиназопозитивный альбинизм), в других случаях этот эффект отсутствует (тирозиназонегативный альбинизм). Известно, что эти два типа альбинизма имеют раздельные локусы генов. При кожно-глазном альбинизме меланоциты определяются, но образование меланосом прерывается на ранних стадиях, поэтому в коже и волосах альбиносов встречаются немногочисленные зрелые меланосомы. Наличествующая тирозиназа функционально неполноценна, она может обеспечить перевод тирозина в ДОФА. Другие варианты кожно-глазного альбинизма включают в себя желтые мутанты, синдром Германского — Пудлака (геморрагический диатез вследствие увеличения числа аномальных тромбоцитов) и синдром Чедиака — Хигаси (возвратные инфекции, гематологические и неврологические нарушения, ранняя смерть при лимфоме). Дефицит меланина при кожно-глазном альбинизме имеет для человека два серьезных последствия: снижение остроты зрения и высокая степень непереносимости солнечного света. Высокая чувствительность человека с альбинизмом к ультрафиолетовым лучам часто приводит к развитию рака на открытых участках кожи. На третьем 10-летии жизни почти у всех живущих в тропиках альбиносов развиваются актинические кератозы или рак кожи. В связи с этим они должны в течение дня пользоваться эффективными местными средствами защиты от солнца и по возможности избегать его воздействия. Сообщается о синдроме врожденного диффузного ослабления пигментации в сочетании с иммунодефицитом. Он сопровождается спленомегалией, нейтропенией, тромбоцитопенией и нарушением функции Т-хелперных клеток. Фенилкетонурия представляет собой нарушение метаболизма фенил аланина, наследуемое как аутосомно-рецессивный признак, при котором в цепи превращений фенилаланина до тирозина заблокировано одно-единственное звено. При этом ослабляется пигментация кожи, волос и радужки. Интенсивность пигментации волос, для которых характерна окраска от очень светлой до темно-коричневой, должна оцениваться только путем сравнения с ее интенсивностью у сибсов, потомков тех же родителей. Меланоциты не изменены, но набор меланосом не полон. Недостаточное образование меланина связано с тем, что избыток в сыворотке и во внеклеточной жидкости фенилаланина и его метаболитов действует как конкурентный ингибитор тирозиназы, блокируя синтез меланина. Витилиго, идиопатически приобретенный ограниченный гипомеланоз, в 30% случаев семейное заболевание, при котором постепенно увеличиваются амеланотические пятна (табл. 51-3). При этом отмечается локальное сегментарное (в пределах одного или более дерматомов) или генерализованное распределение пятен. В некоторых случаях они настолько распространяются, что практически вся кожа становится белой. В типичных случаях пятна витилиго локализуются на разгибательных поверхностях, в местах костных выступов (локтевые, коленные суставы), вокруг мелких суставов кисти, вокруг глаз и рта. В процесс могут оказаться вовлеченными и нижние отделы спины, подмышечные впадины, запястья. Часто он распространяется на кожу гениталий, ладонных и подошвенных поверхностей. В типичных случаях пятна витилиго постепенно увеличиваются в центробежном направлении, появляются новые. Менее чем у 30% ббльных могут спонтанно появляться очаги слабой репигментации, особенно на открытых участках кожи. Волосы в зоне пятен витилиго обычно белые, но могут быть нормального цвета. Большинство лиц с витилиго в целом здоровы, у других с увеличенной частотой обнаруживают заболевания щитовидной железы, сахарный диабет, аддисонову болезнь и пернициозную анемию. Действительно, гипертиреоз, тиреоидит, гипотиреоз и нетиреотоксический зоб в качестве типичного сопутствующего заболевания встречаются при витилиго у лиц в возрасте старше 50 лет, особенно это относится к гипотиреозу. Есть сообщения о синдромах с множественными эндокринопатиями, гипертиреозом, гипопаратиреозом, аддисоновой болезнью, хроническим кандидозом слизистых оболочек и кожи, гнездной алопецией. Более чем у 10% больных может развиться ирит. Вопрос о патогенезе витилиго не решен, согласно классическим представлениям его связывают с разрушением меланоцитов, токсическими предшественниками меланина или лимфоцитами. По некоторым данным, при витилиго обнаруживают антитела к нормальным меланоцитам. Таблица 51-3. Нарушения при ограниченных гипомеланозах типа витилиго Генетические нарушения
Химическое воздействие
Опухоли
Инфекции
Идиопатические
Лечебные мероприятия при витилиго заключаются в защите кожи (экранирование) от воздействия солнечных лучей, косметических методах защиты ре-или депигментации. Более чем у половины больных, леченных с помощью ПУФЛ-А фотохимиотерапии, псораленами и облучением УФЛ-А (солнечный свет или искусственные источники), отмечается существенная репигментация, прежде всего на лице и шее и, кроме того, на туловище, конечностях. Вместе с тем необходимое число сеансов может достигать 200 и более. У некоторых лиц престарелого возраста с обширными зонами депигментации более рациональным и эффективным оказывается местное применение монобензилэфира гидрохинона (бенохин), после воздействия которого происходит необратимая депигментация участков кожи, остающихся пигментированными. Внешне эти больные выглядят вполне здоровыми, но они должны постоянно пользоваться защитными лосьонами. Неполный альбинизм — врожденный, наследуемый по аутосомно-доминантному типу стойкий ограниченный гипомеланоз, сходен с витилиго, но отличается от него распределением пятен и стабильностью во времени, не прогрессирует, выраженность его не изменяется. Депигментированные пятна локализуются на ограниченных участках на руках и ногах и на коже грудной клетки. Для этого состояния типичны белые пряди волос на голове спереди. Цвет глаз в пределах нормы, во всех других отношениях эти лица здоровы. Туберозный склероз, наследуемый по аутосомно-доминантному признаку, у 98% больных проявляется врожденными ограниченными белыми пятнами на коже. Для него типичны также появление примерно к 4-му году жизни припадков, отставание умственного развития и сальная аденома. Белые пятна локализуются на туловище или ягодицах, они отличаются малым количеством меланина, овальной, овально-вытянутой или полигональной формой, напоминаю щей отпечаток I пальца; их число варьирует от 3 до 100. Наиболее типично пятно овально-вытянутой формы или напоминающее, по форме лист американской рябины размером обычно менее 3 см в длину не чисто белого цвета. Пятна ориентированы в поперечном направлении на туловище, на руках и ногах — вдоль их оси. С течением времени их размер и цвет не претерпевают изменений. При гистологическом исследовании в них определяют меланоциты с уменьшенным количеством небольших меланосом. Три ограниченных пятна или более заставляют думать о туберозном склерозе. Для визуализации измененных очагов часто необходимо исследование с помощью лампы Вуда. Для исключения туберозного склероза обследовать с ее помощью необходимо всех лиц с припадками неясной природы или отставанием умственного развития. Кроме того, для определения локализации, размеров и формы кальцификатов и внутрижелудочковых узелков необходимо обследовать родителей и остальных их детей с помощью КТ-сканирования и метода магнитного резонанса. Эти методы могут облегчить диагностику солидных опухолей. Нейрофиброматоз (болезнь Реклингхаузена), наследуемый по аутосомно-доминантному типу, проявляется чаще всего у ребенка в возрасте около 3 лет. Вначале на туловище и руках и ногах появляются множественные бледные желто-коричневые пятна или пятнышки цвета кофе с молоком, диаметр которых может быть менее 1 см или более 15 см. Возможна и генерализованная точечная пигментация или веснушчатость в подмышечных областях. В конце первого и на втором 10-летии жизни часто появляются единичные или множественные мягкие закругленные конусовидные или отвисающие кожные опухоли, покрытые здоровой кожей. Для нейрофиброматоза у взрослого типичны шесть и более пятнышек цвета кофе с молоком, равномерно гипермеланозные, ограниченные, овальной формы пятна диаметром более 1,5см (почти в 5 раз больше, чем у ребенка, у которого он не достигает 0,5 см). Узелки Лиша (пигментированные гамартомы радужки, обнаруживаемые с помощью щелевой лампы) у больных в возрасте старше 6 лет относятся к диагностическим признакам нейрофиброматоза Реклингхаузена, они отсутствуют при синдроме Олбрайта или двустороннем нейрофиброматозе слухового нерва. При синдроме Олбрайта (полиостозная фиброзная дисплазия) число пятен редко превышает 3—4, они локализуются обычно с одной стороны на коже ягодиц или в области шеи. Единичное широкое нейрофиброматозное пятно цвета кофе с молоком неотличимо от пятна при синдроме Олбрайта. Вместе с тем с помощью светового микроскопа можно выявить макроглобулярные скопления меланина во всем эпидермисе, взятом с поверхности пятен при нейрофиброматозе, что обычно не встречается в пигментированных пятнах при болезни Олбрайта и пятнах цвета кофе с молоком, выявляемых у 10% здоровых лиц. Синдром Мойнихена, или «леопарда», наследуемый по аутосомно-доминантному типу, при котором генерализованная веснушчатость (множественные разбросанные небольшие темно-коричневые ограниченные гипермеланозные пятна) сочетается с изменениями на ЭКГ, а в наиболее полном виде и с другими отклонениями (веснушчатость, деформация глаза, стеноз легочной артерии, аномалии гениталий, отставание в росте и глухота). При синдроме Пейтца — Егерса, наследуемом по аутосомно-доминантному типу, появляются гиперпигментированные пятна на губах и слизистой оболочке полости рта от коричневого до синего цвета, сочетающиеся со сходными изменениями кожи и полипами желудочно-кишечного тракта. В отличие от гиперпигментированных участков слизистой оболочки полости рта кожные пятна с возрастом не меняют интенсивности окраски. Это заболевание обсуждается в соответствующем разделе.
Метаболические факторы. Генерализованный коричневый гипермеланоз кожи — типичный признак гемохроматоза, поздней кожной гематопорфирии; пестрой порфирии. Определяемая при гемохроматозе гиперпигментация характеризуется серовато-коричневым или коричневым цветом и неотличима от таковой при болезни Аддисона. Диагноз может быть установлен по данным биопсии кожи, при которой выявляются отложения гемосидерина в потовых железах и отложения меланина. Поздняя кожная порфирия может быть определена клинически по везикулам, пузырям, атрофическим пятнам, склеродермоидным изменениям и просовидной сыпи на коже тыльной поверхности кистей и на лице, а также лабораторным путём no красной флюоресценции подкисленной мочи или по увеличенному содержанию в ней уропорфирина (обычно соотношение уропорфирина и копропорфирина бывает более 3:1). Сходные изменения встречаются иногда и пёстрой порфирии с отчётливым порфириновым профилем с особо высоким уровнем фекального протопорфирина. Кожные изменения при этом идентичны гаковым при поздней кожной порфирии. дифференцировать их следует на основании различий в реакции на лечение и связанных с этим медицинских проблем. Гиперпигментация здоровой кожи на открытых участках тела может быть обусловлена первичным билиарным циррозом. Хроническая почечная недостаточность также может сопровождаться диффузной гиперпигментацией. Факторы питания. При хроническом дефиците питания появляются гиперпигментированные пятна грязно-коричневого цвета, особенно на коже туловища. При некоторых дефицитах избирательного характера, например при белковой недостаточности, сопровождающей квашиоркор, или при потере белка, что происходит при хроническом нефрозе, язвенном колите и синдроме мальабсорбции, иногда уменьшается интенсивность окраски волос, которые становятся красновато-коричневыми и в конечном итоге седыми. При других болезнях, например спpy, возможен коричневый гипермеланоз в любой части тела. При пеллагре же зона пигментации ограничивается участками кожи, подверженными воздействию света или травматизации. Дефицит витамина B12 сопровождается преждевременным поседением волос и гипермеланозом, особенно отчетливо выраженным вокруг мелких суставов кистей Пигментная крапивница отличается множественными неправильной округло-овальной формы пятнами и папулами от желтовато-коричневого до красновато-коричневого цвета, что связано с наличием меланина в эпидермисе над скоплениями тучных клеток. Энергичное растирание таких зон приводит к появлению уртикарной сыпи (признак Дарье). У детей кожные изменения появляются обычно в период раннего детства и часто спонтанно исчезают через несколько лет, Течение заболевания обычно доброкачественное, но у 30% больных появляются гиперемия, зуд, крапивница, менее чем у 15% наступают рвота, глубокие обмороки, шок. Эти проявления, вероятно, связаны с высвобождением тучными клетками гистамина и часто сочетаются с усиленной экскрецией с мочой свободного гистамина и его метаболитов. Уровень 5-гидроксииндолуксусной кислоты в моче находится в пределах нормы. Антигистаминные препараты малоэффективны. Системный мастоцитоз, при котором тучные клетки диффузно инфильтрируют печень, селезенку, желудочно-кишечный тракт и кости, относится к редким состояниям. Иногда развивается тучноклеточный лейкоз. Неизвестные факторы. Генерализованный коричневый гипермеланоз типа гипермеланоза при аддисоновой болезни может быть связан с системной склеродермией на ее ранних стадиях. Изредка генерализованная гицерпигментация развивается у больных с хронической печеночной недостаточностью, особенно в результате портального цирроза. Патогенез пигментации при том и другом состояниях неизвестен, уровень меланинстимулирующего гормона не повышен. При синдроме Кронкайта—Канада приобретенные лентигоподобные коричневые пятна сочетаются с полипозом желудочно-кишечного тракта, начиная от желудка и до прямой кишки. За несколько месяцев до появления кожных изменений обычно начинаются диарея, боли в животе, уменьшается масса тела. Гипопигментированные пятна в сочетании с перифолликулярными гиперпигментированными пятнами встречаются при склеродермии; эти пятна лишены меланоцитов. Гипомеланозные пятна могут обнаруживаться и у небольшой части больных саркоидозом. В этом случае цвет пятен не чисто белый, их границы нечетки. Они могут локализоваться над дермальными узелками, особенно на руках и ногах, а иногда и на туловище.
|
 |